 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 
CNDA上周(7月24日)颁布了“关于调整药物临床试验审评审批程序的公告(2018年50号令)”,有些意料之中,也有些突然。 去年发布征求意见稿后,经过7个月出台正式公告,与其他征求意见稿相比,本次公告的效率颇高,为国家局点赞!
猫老师的夏天很忙,好久未提笔写作文,至此大事之际,虽然无法争分夺秒第一时间发布解析,还是执意提笔颂扬一下CNDA的开拓创新的改革力度!
开卷有益,我们一起在火热的夏日速读“6大看点”
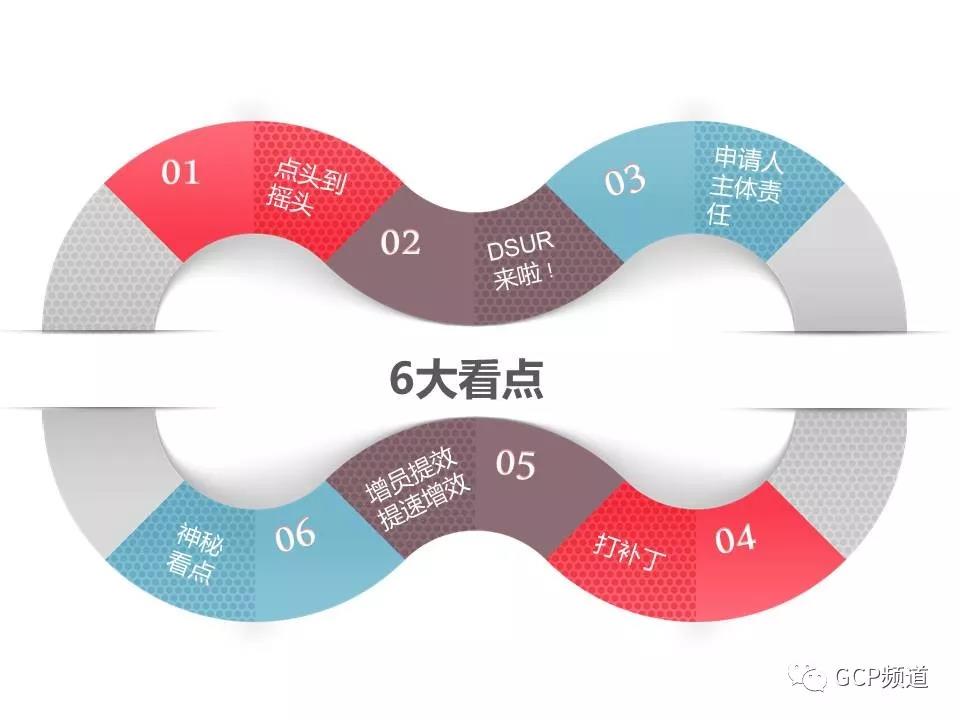 1.“点头”制改为“摇头”制
1.“点头”制改为“摇头”制
众所周知,FDA对临床试验申请实行较为宽松的审批制度,在收到新药试验申请30日内,申报者如未收到任何有异议通知时,新药试验即可自行开始。多年以来,我们国家一直采用对临床试验申请实行较为严格的审批制度,而且以颁发临床试验批件作为批准临床试验的方式,审批流程也很长,这也就是所谓的“点头”许可制度。本次公告突破性地将“点头”制改革为现在的“摇头”制,即不用再“点头”,而是对审核不同意的申请“摇头”。从“点头”制到“摇头”制的变迁,实质上是药物研发审评审批能力的提升和制度成熟的直接体现。
“摇头”制除了体现在本次公告的CTP的审批方面,也同时体现在临床试验机构备案制的实施方面。
“摇头”制的默许审批是具有革命性的变革!为CNDA的“摇头”制点赞!
2.明确提出DSUR的要求
ICH E2F明确提出了药物研发期间安全性更新报告(DSUR)的要求,我们国家既往对于PSUR有明确要求,而DSRU的要求不清晰。本次公告提出,申请人在获得首次临床试验许可后,应至少每年定期向CDE提供DSUR,这也是我们国家首次明确了提交DSUR的要求。同时,对于申请人需要在药物研发期间的安全性更新报告的内容也做了以下要求,虽然仅仅是简单的几句话,但是也是巨大的监管变革。简单没关系,我们可以参考ICH E2F详细要求,我们中国也是加入了ICH嘛!为国家局采纳DSUR,强化药物研发过程中的管理点赞!
全球研发和上市状况
正在进行中和已完成的临床试验
新增的安全性结果
重大生产变更
整体安全性评估
重要风险总结
获益-风险评估
下一年总体研究计划等
公告中也规定DSUR的上报时间(参见以下4条),规定DSUR周期的起始日时在获得首次临床试验许可为时间点,不知这里的“首次临床试验许可”是指我们CNDA的许可,还是指ICH E2F中采用的研发国际诞生日(DIBD)?猫老师和大家一起搬个板凳期待官方的解读^
在获得首次临床试验许可后开始。
一般每年一次,于药物临床试验许可后每满一年后的二个月内提交。
CDE可以根据审查情况,要求申请人调整报告周期。
逾期未提交的,申请人应暂停药物临床试验。
3.落实申请人的研发主体责任
默认许可是所谓的“大批件”的形式,而不用每个分期的临床试验都要做一次申请。同时,本次公告特别说明了满足以下4个特定条件,申请人可不经沟通交流直接提出临床试验申请,简化流程的同时,进一步强调了申请人的主体责任。
技术指南明确、药物临床试验有成熟研究经验。
申请人能够保障申报资料质量的。
国际同步研发的国际多中心临床试验申请。
在监管体系完善的国家和地区已经获准实施临床试验。
谁申请,谁承担主体责任!公告强调了申请人重任扛起来,点赞!
4.继续“打补丁”改进中提升
近年来,我国监管机构优化药物审评审批改革、鼓励创新,先后修订和出台了很多法规和公告。本次50号公告依旧采用“打补丁”的形式,明确说明本公告自发布之日(2018年7月24日)起实施,此前与本公告不一致的以本公告为准。
“打补丁”是步伐大,频率高的变革过程中常用的有效做法。在原有的基础上,推陈出新,提高变革速度和效率,点赞!
5.CDE增员提效,提速增效
CDE在大幅度扩充审评员队伍的规模,我们也常在官网上看到审评员招聘的通知。优化审评审批流程,CDE也将压力留给了自己。我们也应该伸出大拇指为国家局增员提效的变革点赞!
自50号公告颁布之日起,载入历史史册的获得<药物临床试验批件>的法规规定的时间为175/155天(斜线前为一般审批时限,斜线后为特殊审批时限),而且这个时间只能说是法规上的时限,实际执行中偏差很大。本次公告明确了在我国申报药物临床试验的,自申请受理并缴费之日起60日,申请人未收到CDE否定或质疑意见的,可按照提交的方案开展药物临床试验。这是时限是非常清晰了,到了60日大限,也无需再继续苦苦等待了,CDE不“摇头”,申请人就可以满心欢喜的开始了。
当然,究竟多久可以获得默许药物临床试验许可,也取决于“pre-IND meeting”沟通交流会议的日期, 这个日期可是需要双方协商的。
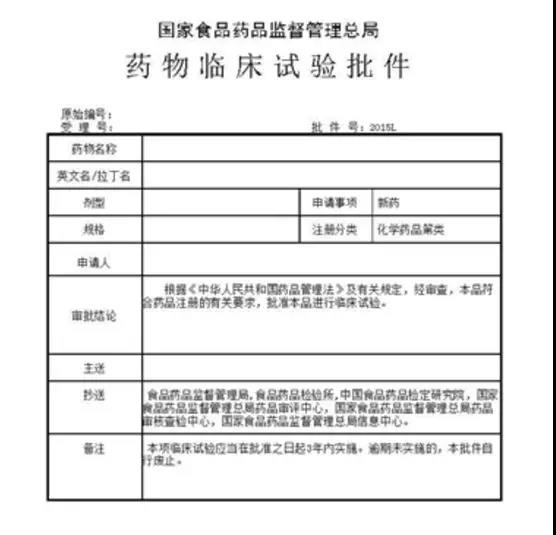
6.最后看点!再看一眼值得纪念的药物临床试验批件!
猫老师在文末,特别附上一张CTP。因为我们更应该为GCP征途上,这张不朽的证书点赞!
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx


 PHEXCOM公众号
PHEXCOM公众号