 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务
English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 English [切换]
药用原辅包质量审计、注册及供应;GMP 质量体系咨询;药物制剂研发技术支持和信息服务 昨日,国家食品药品监督管理总局发布了<国家食品药品监督管理总局关于征求加快解决药品注册申请积压问题的若干政策意见的公告>(2015年第140号)”(以下简称“公告”),向社会公开征求意见。
力图解决积压难题
针对药品注册申请大量积压的窘状,2015年的前7个月,国家总局与药审中心(CDE)采取了一系列举措。逐一点评如下:
1) 去年底,CDE大量招聘审评员。
点评:早就该这样做了!因为以上问题早在3年前就已十分突出,每年“两会”,制药行业代表均会提出。
2) 借调外省人员帮忙审评。
点评:该办法对于解决药品注册申请积压问题可以起到一定作用,但解决不了大问题,非长久之计;同时,借调人员的专业水平也值得商榷。
3) 计划购买“第三方服务”。
点评:这一点不太现实。单不论“第三方服务”的专业水准,光是“打擦边球、甚至暗度陈仓”的行径很快便会成为“潜规则”。
4) 建立<立卷审查标准>。
点评:这是个好办法!但令人遗憾的是:该标准是内部标准、保密、不对外公开。何不将标准“张榜公示”,明确告知企业达到这些要求后再来申报,以发挥“该标准延缓申报时间、减少申报数量”的作用?
5) 5月27日,国家总局发布<药品、医疗器械产品注册收费标准和实施细则>,大幅提高申报费用。
点评:此举是想通过提高费用门槛,迫使那些财力不雄厚的研发单位减少申报数量。但笔者认为作用有限,反而可能会增加企业负担,还不如让企业将这笔资金投入到研发中,用心将产品做好。
6) 7月22日,国家总局颁布<关于开展药物临床试验数据自查核查工作的公告>(2015年第117号)。
点评:赞一个!“临床试验数据的粉饰甚至造假”早已是业内多年来的“潜规则”。总之,极少听到有失败的临床试验便可知晓其中的问题多么严重。结果是:到了临床,相当一部分药物成为“安全无效”的产品,令临床医生对国产药信心不足。
7) 7月31日,国家食品药品监督管理总局发布<关于征求加快解决药品注册申请积压问题的若干政策意见的公告>(2015年第140号)。
点评:非常好!其中透露出很多积极信号。至于这些措施能发挥作用几何,让我们拭目以待!
意见建议及日本经验
在提出建议之前,先打个比方:某效益极佳的单位向社会公开招聘,应聘者肯定趋之若鹜。该公司张贴出的招聘广告有两种。第一种:男性、年龄,仅此两项;第二种:男性、年龄、政治面貌、学历、工作经历、外语水平等等。如上两种情形,应聘者的数量必将迥然不同,单位人事部门与领导的面试工作量也就天壤之别了。
本人12年前曾在日本国家药品检验所药品部学习进修,其时该部门仅10人,需面对全国1600家企业申报来的品种,他们是如何应付自如的呢?
一、公示出“各剂型的关键性指标要求”
即制订出每一剂型的关键性评价指标与申报要求后张榜公示,研发单位做到后方可申报,否则不要来。以口服固体制剂仿制药研发要求为例:
1.连续3批、每批至少10万片。
“3批”代表工艺与产品的稳定性,“10万片”代表工业药剂学。如存在无法满足该要求的特例,请提前与该部来信协商,确定被允许后再开始研发,以免造成浪费。
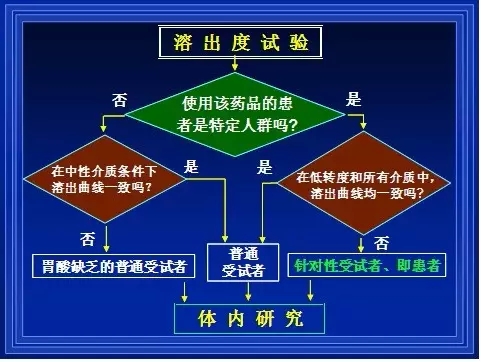
2.在有针对性的溶出度试验条件下,批批样品的多条溶出曲线均需与原研制剂一致。
该针对性的溶出度试验条件由该部门专家制定,给出具体的试验办法与操作流程,即<日本口服固体制剂生物等效性试验指导原则>中的溶出度试验部分,该规定是针对大量已上市原研制剂多条溶出曲线剖析结果,总结归纳、演绎推理所得,既包含体内外相关性,又包含针对处方筛选和工艺开发的区分力,因为该部门的专家们同时也审阅了大量原研制剂申报材料,从中找到了规律。
该部门甚至做了一件更有意义的事情:由国家主持、测得“原研制剂多条特征溶出曲线”予以公布,研发时与市场抽验时均需遵循。如此,研发单位无需再剖析,以免造成剖析结果的五花八门、仿制制剂开发途径的千奇百怪。
笔者建议:我国药监部门应尽快成立“国家队”,测定并建立起<原研制剂多条特征溶出曲线数据库>;并应尽快改变目前仅复核“三个一”(即“一个介质、一个时间点、一个限度”)的现状,CDE应请各省级药检所帮助复核多条溶出曲线。
3. 生物等效性试验(BE试验)的样品应至少是申报时的生产规模或更大。
受试者不能仅开展年轻男性的体内生物利用度检测,还应根据不同药物的特性,酌情扩大年龄和性别等。
当申报企业提出“我公司仿制制剂虽然体外溶出行为与原研制剂不一致,但有把握体内生物利用度一致,不愿再完善制剂研发”时,日本规定“体外溶出度试验与体内生物利用度的相关性”如下:
4.大生产。
如是在BE试验样品生产规模的10倍以内,可依靠体外溶出曲线对比来“确保大生产样品的质量与BE实验时的样品质量一致”,或是与原研制剂做比较,多条溶出曲线应一致。
5.上市后的市场抽查。
日本国家和地方药检所仅抽查“多条溶出曲线”。该条规定犹如“紧箍咒”,迫使生产企业深入研发和控制好制剂的各个要素,如此才能确保每批样品的多条溶出曲线保持恒定,以此来保证每批样品的临床有效性和均一稳定性。
二、 公示出“各品种的关键性指标要求”
启动某品种审评后,在审阅最初的3~5家申报材料后,即刻在CDE网站上公布出该品种的具体技术要求与核心要素,随后“排队当中达不到上述要求的请主动撤审”和“尚未申报、正在研发的单位可有的放矢地进行研究”。
三、 唯有通过一针见血、切中要害的技术要求才能迫使企业进行深入研发,从而起到延缓申报时间、减少申报数量的效果,并将企业有限的资金用到研发实处。
总结:笔者认为出现大量积压的根本原因是,我国CDE公布的技术要求很多较为模糊,导致研发单位都觉得“自己行_花费不多就能跨过CDE设定的技术门槛”,所以都信心满满、蜂拥而至。
反观发达国家:一条“10万片”的要求就可使多少不具备大型生产设备的研发机构望洋兴叹,可以使多少财力不足的研发机构折戟沉沙,因为对于那些制剂含金量高的品种(如难溶性药物、肠溶制剂、缓控释制剂、pH值依赖性制剂、治疗床狭窄药物制剂等),要做到在各介质中的溶出行为均与原研制剂一致是要付出相当大的代价与精力的。
最后,笔者寄望:我国CDE的审评人员应尽快提升自我专业水准,要深谙药物研发的核心要素与关键流程,因为“只有高水准的教练员/裁判员才会产生高水准的运动员”,这是本行业发展的客观现实与规律。
【编辑:amanda】 国际药物制剂网 本文链接: http://www.phexcom.cn/hydt.aspx


 PHEXCOM公众号
PHEXCOM公众号